

What We Stand For?
Health has never been distributed equally, and we’re here to change that. We support physicians, students, and advocates with the tools to lead on equity, uplift evidence, and center the communities most impacted by injustice.
Explore Our Mission.webp)
.webp)
.webp)
Take Action Today Be The Change
Whether you’re a doctor, student, or advocate - there’s a place for you here.



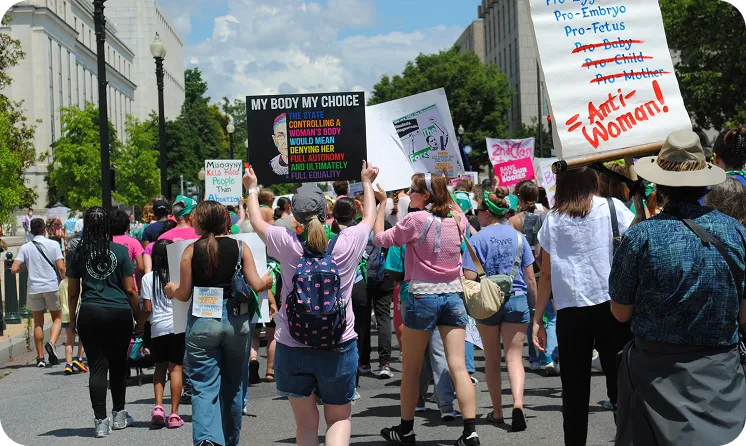


Advocacy and Impact Backed by Action
.webp)
meetings with Congressand Health Agencies
Impact Stories Shared
Press releases and statements

Join the Movement, Wherever You Are
Whether you’re new to advocacy or a seasoned voice for change, our virtual and in-person events offer meaningful ways to connect, learn, and lead - on your schedule.


.webp)
Where We Take a Stand
We lead on the policies that matter most - grounded in evidence, informed by clinicians, and focused on outcomes that advance equity and justice for all.
.webp)
.webp)
.webp)
Real Stories. Real Impact.
Meet the physicians, students, and advocates turning frontline experience into policy leadership - one story, one action at a time.